
Selection, Concatenation and Fusion of Sequences
|
Selection, Concatenation and Fusion of Sequences |
| < Comand line mode | Table of content | Output Files > |
|
To run SCaFoS To choose the usage To make an OTUs file To select files with chosen species To create datasets To obtain help To run SCaFoS To choose the usage To make an OTUs file To select files with chosen species To create datasets To obtain help To run SCaFoS To choose the usage To make an OTUs file To select files with chosen species To create datasets To obtain help To run SCaFoS To choose the usage To make an OTUs file To select files with chosen species To create datasets To obtain help To run SCaFoS To choose the usage To make an OTUs file To select files with chosen species To create datasets To obtain help To run SCaFoS To choose the usage To make an OTUs file To select files with chosen species To create datasets To obtain help To run SCaFoS To choose the usage To make an OTUs file To select files with chosen species To create datasets To obtain help To run SCaFoS To choose the usage To make an OTUs file To select files with chosen species To create datasets To obtain help |
Graphical mode
Under a Linux system, the program is loaded by typing:
scafos
or the following command in interpreted mode: perl scafosXXX.pl
where XXX is the version number of SCaFoS
In the Windows environment, open a command prompt window and type one of the previous commands to run SCaFoS. In a standard installation of Windows, to open a command prompt window:
In the Mac OS X environment, open a X11 window and type one of the previous commands run SCaFoS. The X11 windows are accessible in the Applications>Utilities folder. For all operating systems, be sure that the scafos environment variable is defined previously to run SCaFoS (see the instructions for installation section) Input and output files are described in Input files format section and Output Files format section respectively.
The three usages are available according to the chronological use of SCaFoS:
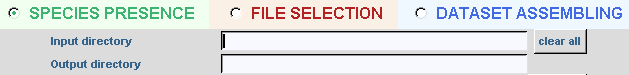 Last options selected are automatically reloaded. By clicking on the clear all button, all options are removed and default values are reloaded. Input and output directories are always needed. If the output directory already exists, you must confirm to replace the old one or type a new name. For each usage, some parameters are needed or optional and only potential parameters will be activated; the description of these options will follow. Once the parameters are chosen, a simple click on RUN button executes the program. A new window will appear while the application is running. Close the result window, by clicking on the CLOSE button; the user can make another analysis by changing options or quit by clicking on the QUIT button. With the VERBOSE option, a detailed description of the run is displayed. In all following explanations, out will be used to refer to the output directory.
To create an OTU file, first choose the
SPECIES PRESENCE radiobutton.
The following options are available:
 It is possible to continue with a concatenation according to the length of sequences. The program prompts the user for this option: a concatenated sequence will be made for all species found at least once in the aligned sequences files. The concatenated file resulting of this rough concatenation will give a first draft of phylogeny because this dataset contains many missing data and lacks good choice of paralogous sequences
Choose radiobutton SELECT FILES.
Since SCaFoS will perform sequences selection, it's necessary to indicate the OTUs file. To easily select OTUs file, click on the browserbutton to open a files selection window. You could automatically eliminate sequences that are too short by typing the Minimal length to choose a sequence. By default sequences with a length shorter than 10 percent of the longest sequence are removed from the analysis and are not taken into account to calculate the evolutionary distance.  According to the checked option, selected files of aligned sequences are written in output directory; three options are available:
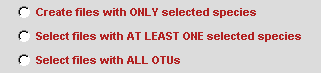 Except for the first option for which unselected sequences are removed from the file, the selected aligned sequences files are identical to the initial files
This step is the central point of SCaFoS because it generates a lot of
information that could be used to choose genes and species, so several
options are associated with this usage for which the user needs to
click the radiobutton DATASETS ASSEMBLING.
This usage needs to choose an OTU file. Selection criteria
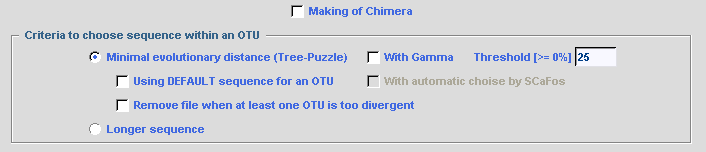
When MAKING of CHIMERA is chosen, if no complete
sequence exists for an OTU, a new sequence is created with portions from
different species within the OTU (remember that with the
MINIMAL LENGTH option is applied on the created chimera).
Several criteria are used to determine the best sequence within an OTU:
 Selection of default sequences
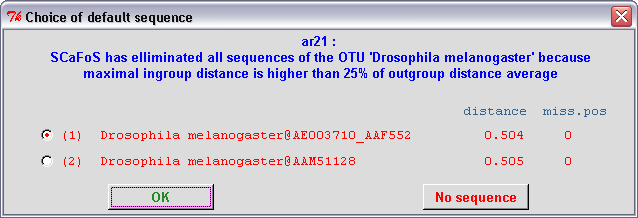
In the semi-automatic mode, when SCaFoS is unable to select an unique sequence for the OTU
(i.e. when Using DEFAULT sequence for an OTU option
is selected), the user must validate the choice of the sequence guided
by the evolutionary distance, the amount of missing positions, and
eventually the deviation in composition. Remember that the list
of sequences is written in red when at least one sequence is too divergent,
otherwise the list is written in blue. Three options are available:
If all sequences are identical, SCaFoS automatically selects the first sequence without asking the user. Sequence selection in semi-automatic mode could be very time comsuming, but selection of default sequences is a real gain when the same dataset is use for various purposes with minor variations in definition of OTUs. After closing SCaFoS, copy the .def files newly created from the output directory to the input directory; on the next run, the previously selected sequences will be used and no question will be asked to the user for these OTUs. Creation of subdirectories
To minimize frequency of missing characters in final data set, the user can
create subdirectories with selected files according to:
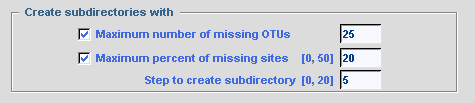 Global options
The value given for the option Maximal percent of
missing sites corresponding to complete sequence determines the
threshold for which a sequence is treated as complete and not included in
the chimerical sequence. This option is used only if the
MAKING of CHIMERA option is checked; otherwise all
sequences are taken into account to calculate the evolutionary distance
independently of their completeness.
To obtain a phylogenetic tree where sequence names are written in different colors according to their selection by SCaFoS, check the corresponding option: Tree in postscript format colored according to selected sequence In complementary of computing of evolutionary distances, it is possible to display the variation in sequence composition towards the average composition of all sequences in the file. By checking the option: Determine variation in sequence composition, this information is displayed during the interactive choice by the user and in various output files. 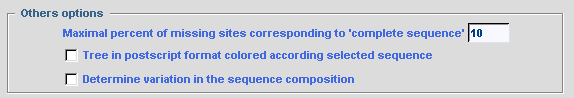 The concatenated file can be output in several formats: FASTA, PHYLIP in sequential format, MUST and NEXUS. Just check the desired options. Commands can be automatically added to the nexus file to obtain output files suitable for PAUP or MrBayes. The user can also modify these optional commands or add his/her own commands in the NEXUS file with the for other option: a text editor will be displayed to type appropriate commands. Be careful because no verification for the validity of these commands is made by SCaFoS. For all formats it is possible to choose the order of the concatenated sequences with the Output order option. These options are also used for the concatenated files in subdirectories if needed. Checking the Add number of characters to the OTU name adds the length of the concatenated sequence at the end of each OTU name: i.e. the number of real characters. 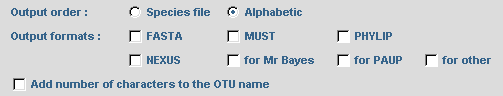
A short help for each option is displayed when the mouse is motionless over the
corresponding option. A global help is displayed by clicking on the
HELP button. In the new opened window, it is possible
to display the full online help available directly on this web site.
|
| < Comand line mode | Table of content | Output Files > |