Created by: Liisa B. Koski
Contact email: autofact@bch.umontreal.caPlease send email if you wish to be kept updated on AutoFACT improvments.
To unpack the file type 'tar xvf AutoFACT_v3_4.tar'
Koski LB, Gray MW, Lang BF and Burger G. AutoFACT: An Automatic Functional Annotation and Classification Tool.
BMC Bioinformatics. 2005 Jun 16;6(1):151
1. Analyzes nucleotide and protein sequence data.
2. Determins the most informative functional description by combining multiple blast reports from several user selected databases.
3. Assigns putative metabolic pathways, functional classes, enzyme classes, GeneOntology terms and locus names.
4. Generates output in HTML, text and GFF formats for the users convenience.
5. Performs transitive EST annotation (trEST), see below for details (NEW to version 3.2).
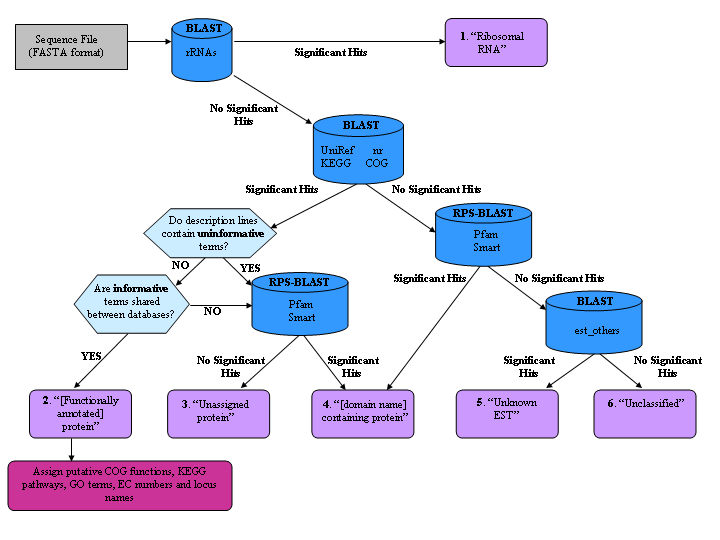
AutoFACT takes a single FASTA formatted sequence file as input and proceeds to assign each sequence to one of six annotation classes:
Step 1. Each sequence is BLASTed against the users choice of the following databases:
Step 2. When there is a significant hit (based on user specified Hit Length and % sequence identity cutoffs) to one of the ribosomal RNA databases (LSU or SSU) the sequence is immediately classified as a Ribosomal RNA.
Step 3. If the sequence is not a ribosomal RNA, sescription lines of significant hits to the remaining databases (based on a user specified E-value cutoff) are examined for the presence of functionally uninformative terms such as 'hypothetical', 'unknown', 'chromosome', etc. In the case of a match, the next hit is scrutinized and so on until a description line without uninformative terms is found, for example 'proton-transporting ATP synthase'. This hit is then taken as the 'informative hit'.
Step 4. A search for common terms between the informative hits from each database is performed. A database order of importance for the annotation transfer is user specified, so that informative terms from the first database are searched against informative terms from the remaining databases in the given order. If a match is found for a least one informative term, the description the description line from that database is assigned to the new sequence.
Step 5. When informative terms are not shared between any of the databases or when only uninformative hits are found, the sequence is checked for the presence of protein domains. If the sequence contains a protein domain it is classified either as a [domain-name] containing protein or a multi-domain containing protein.
Step 6. If the sequence contains no domain it is classified simply as an Unassigned protein.
Step 7. A sequence is also classified as a [domain-name] containing protein when the only significant hit is to one of the domain databases.
Step 8. In the absence of a domain and when annotating ESTs, if a significant match is found to an entry in est_others the sequence is classified as an Unknown EST.
Step 9. Otherwise the sequence is Unclassified.
Thanks to the suggestion from Susan Renn (SRenn@CGR.Harvard.edu) we have added Transitive EST annotation to the AutoFACT pipeline. This step is invoked by using the -t flag on the command line. If an EST is initially annotated as 'Unknown EST' (i.e. it's only hit is to the est_others database), AutoFACT will take the full length est_others sequence and run that sequence through the AutoFACT pipeline. If the est_others sequence can be assigned a functionally informative description then this description is transitively assigned to the initial EST sequence. In some instances this can greatly improve the annotation information. When an annotation is assigned via the transitive EST, it's description begins with 'trEST; annotation name'.
Rickettsia prowazekii [GeneQuiz]
Saccharomyces cerevisiae [PEDANT]